
A Bioinformatics Pipeline Powered by
Deep Machine Learning for Virology InvestigationsNovel pathogen-specific analytics and mining of Next Generation Sequence data.
- Full-service analytics
- Pre-built tools/algorithms
- Custom development
- Analysis and writing services
Now hiring for development of novel quasispecies analytic pipeline for Hepatitis B Virus (HBV)
GATACA
GATACA is developing novel software solutions for virologists. Funded by Small Business Innovation Research (SBIR) awards from the National Science Foundation (NSF) and the National Institutes of Health (NIH/NIAID), our current focus is on hepatitis B virus (HBV) bioinformatics aided by deep machine learning (ML). Our algorithm, GAT is a novel framework for de novo assembly of HBV NGS output into quasispecies that we are now empowering with artificial intelligence (AI)—specifically ML methods to learn the ‘syntax’ of the infecting strains. GAT/ML™ trained on HBV variation using our proprietary database benchmarked as superior against leading viral assemblers in simulation studies and tests on patient samples, and—is currently undergoing further optimization for enhanced analytics of patient samples. Our abstract on a soon-to-be published patient case-study demonstrating GAT/ML’s utility to identify rare variants and quasispecies will be presented at EASL (London, August 20201).
Other development underway includes statistical modeling of quasispecies evolution, and prediction models to forecast disease progression.
Applications for GAT/ML™ are in labs using NGS for virology discovery
- Academic scientists in research and in multidisciplinary translational settings, to eliminate duplication, increase time to publication, pinpoint problems and maximize grant resources.
- Antiviral drug developers screening drug candidates for emerging resistance, enabling early ‘kill decisions’ to avoid high-risk compound advancement.
- Clinical scientists running trials on vetted drugs, to improve patient selection and perform on-trial monitoring.
GAT/ML™ trained on a vast knowledge base of HBV variations processes a high NGS volume and profiles existing dangerous HBV quasispecies with speed, precision and unprecedented reproducibility.
Analytics for HBV and HCV are ongoing in-house. GAT/ML™ v1.0 for HBV will be available for testing in Summer, 2020.
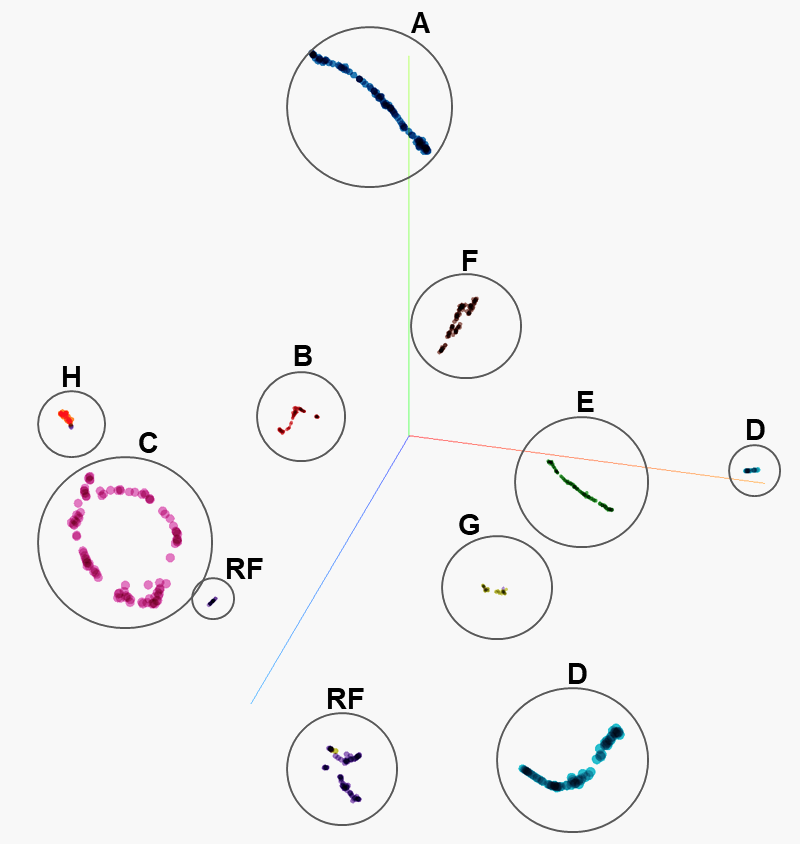
Using our proprietary first ML module trained on HBV variation, the sequences clustered into distinct major genotypes and subtypes, focused GAT assemblies and improved haplotype accuracy
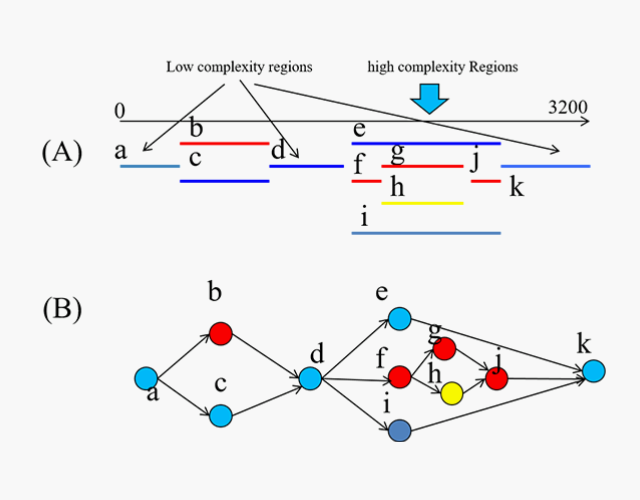
Our Pipeline for HBV—Available Soon for Direct Access
GAT/ML™ for resolving HBV quasispecies from NGS
Assembly Tool for HBV genomes
- QC of NGS, including removal of non-target reads
- Novel de novoassembly method, interspersed with multiple cooperative ML modules that focus and refine assembly, and predict variants
- Proprietary database housing all HBV variation, including strains unrecoverable from public repositories
- Identification of viral strains at resolutions previously not possible, including unknown / poorly documented complex variants
- Identification of variants in overlapping reading frames (e.g., HBsAg and Reverse Transcriptase overlap in HBV)
- Links variants to progressive phenotypes
- Quasispecies reconstruction and modeling
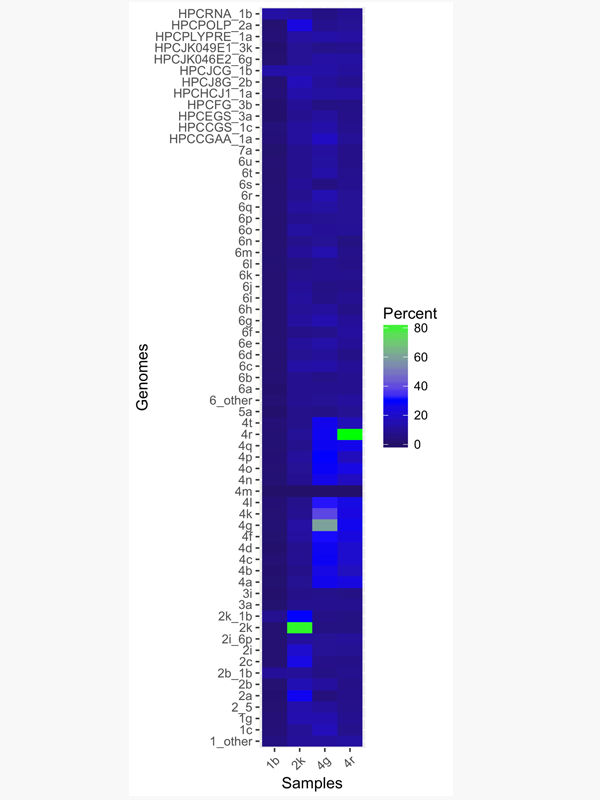
Analysis of HCV and HBV Samples, In-House
- Mutation tracking and ID, heterogeneity quantification and resistance profiling
- Genotyping directly from NGS reads
-
- On datasets with ≥1,000,000 and as few as 1,000 reads
- Quantifies the fraction of each subtype present in the dataset
- Full report, including all variants and regions
Data Quality and Variant Analysis In-House
- QC Error correction
- Extraction of contamination and redundancy
- Genotype and SNP Calling
- Drug Resistance Screening
- Mutation Signature ID
- Annotation
- Resistance Profiling
- File and sequence platform concordance
- Other NGS statistics
Sample Outputs








Our Features
Take control of your virology research and gain more insight faster
- Database Metrics
- Mutation capture
- Genome-specific trait ID
- Dynamic databases
- Integration of workflows
Data Metrics
Baseline quality controls allow removal of unwanted reads (e.g., too short, low quality bases); error correction, extraction of redundant and contaminating reads (e.g., microbial, human). The resulting dataset includes only the viral data, thus streamlining down-stream analytics.
Trait Identification
Key protein coding regions are critical to understanding the specifics of viral structure and mutation potential. Our methods automatically segregate and identify these regions with unprecidented rapidity.
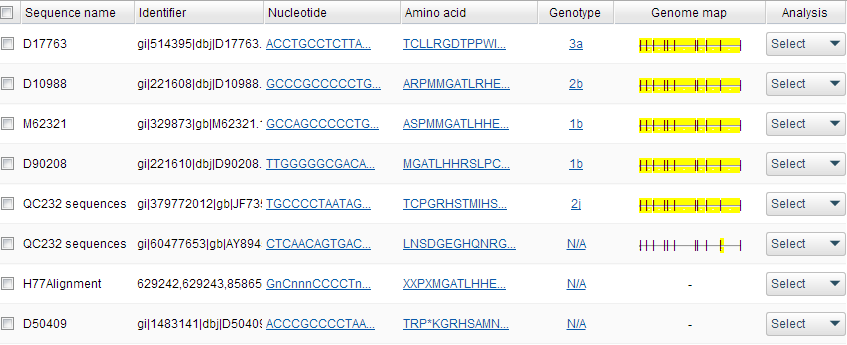
Mutation Capture
Resolving quasispecies composition is a daunting challenge in viral informatics. Our proprietary assembly algorithm utilizes an iterative scheme to produce contigs and interconnections highly representative of intra-host variation, including long-range and in the low frequency realm. Alignments enable comparison of output to our dynamic databases with up-to-date resistance mutations, including DAAs.
Dynamic Databases
Implementing a dynamic design has enabled us to overcome the challenges associated with static database design; our databases are scalable and flexible, require minimal data reconstruction, and are updated regularly with emerging variant information (resistance, replacement, escape, etc).
Integrate Workflow and Experiments
The key to a solid data management system is integrating the workflow, such as attaching annotations to sequences, creating data linkages, extracting sequence-specific features for simplifying and unifying your data input with automation; discerning batch effects and meaningful offsets, and unmasking hidden patterns and trends can be ascertained by integrating the results of multiple experiments with proper normalization and cross-compiling.
USEFUL LINKS

Additional Offerings

Custom Designed Analysis Tools
Strategies and algorithms custom designed for small complex genomes.
- Unique data management and analytic methods that resolve high genetic heterogeneity and allow longitudinal analyses to track mutations and viral evolution
- Custom Analytic Reports
- Custom design of algorithms for other small complex genomes
- Customize existing and new algorithms for existing and emerging technical outputs, including:
-Third-gen sequencing
-Gene expression (RNAseq)
-Protein expression (Cytof)
Notice: Trying to access array offset on value of type null in /opt/bitnami/apps/wordpress/htdocs/wp-content/plugins/kingcomposer/shortcodes/kc_single_image.php on line 74
Notice: Trying to access array offset on value of type null in /opt/bitnami/apps/wordpress/htdocs/wp-content/plugins/kingcomposer/shortcodes/kc_single_image.php on line 78

Editing / Writing Services
Our team of skilled writers and field-matched bioinformatics experts will advise on the strategy, structure, and creation of your scientific documents and presentation materials. Our team can help you maximize the impact of your work by creating clear, concise, and powerful publication- and presentation-ready material. Services are offered at varying levels to match individual needs and a work plan will be created to match.
- Manuscripts
- Grant Preparation
- Creation of SOPs
- Research Reports
- PowerPoints
- Pamphlets and Brochures
- Custom Educational & Promotional Materials
- Services of value to both native and non-native English speakers
Notice: Trying to access array offset on value of type null in /opt/bitnami/apps/wordpress/htdocs/wp-content/plugins/kingcomposer/shortcodes/kc_single_image.php on line 74
Notice: Trying to access array offset on value of type null in /opt/bitnami/apps/wordpress/htdocs/wp-content/plugins/kingcomposer/shortcodes/kc_single_image.php on line 78
Study Design Consultation
Our team has extensive experience working with, educating, and advocating along-side priority populations identified in the Viral Hepatitis Action Plan. We are available and interested in partnering with research teams looking to advance the Action Plan by decreasing health disparities through education, awareness, and improved access to care.
- Health Disparities Research Study Design
- Grant Writing
- Public Education and Advocacy
- Community Awareness
